
 This project has received funding from
the European Union's
This project has received funding from
the European Union's 
Parahydrogen-Induced Polarization
As discussed in previous posts, a source of nuclear spin polarization is required in a ZULF experiment, and one option is parahydrogen-induced polarization (PHIP).
The main drawback of magnetic resonance is the low sensitivity, which is determined by the small differences in state populations which are distributed between the energy levels of a spin system according to the Boltzmann equation at thermal equilibrium. With hyperpolarization we can generate a huge difference in state populations which leads to an enormous increase in the NMR signals. This is illustrated in Figure 1.
Parahydrogen induced polarization is an inexpensive and easy-to-handle hyperpolarization method that can provide high polarization levels on target molecules on a timescale of seconds. In this article we will describe how molecules can be hyperpolarized using PHIP.
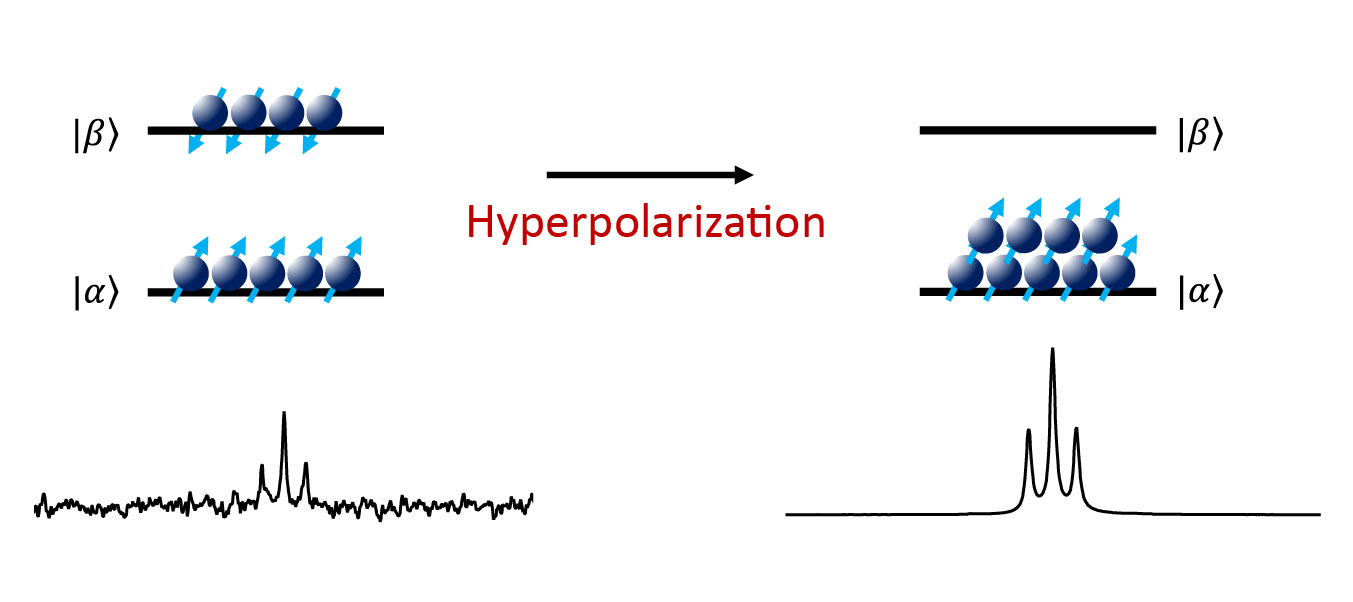
Figure 1: An illustration of nuclear spin hyperpolarization.
Molecular hydrogen (H2) has two nuclear spins, which give rise to four different spin states: one singlet- or para-state, pH2 (the para spin isomer has a total
spin angular momentum I=0, and is called the ’singlet’ isomer because mI=0) and three triplet- or ortho-states, o-H2 (the ortho spin isomer has a total spin angular
momentum I=1 and is called the ‘triplet’ isomer, because the angular momentum projection along the z-axis can take three values; mI=−1, 0, 1). These four states of
the system can be written as linear combinations of Zeeman basis states:
S0 = (|𝛼𝛽⟩-|𝛽𝛼⟩)/√2
T+1 = |𝛼𝛼⟩
T0 = (|𝛼𝛽⟩+|𝛽𝛼⟩)/√2
T-1 = |𝛽𝛽⟩
where S and T are singlet and triplet states.
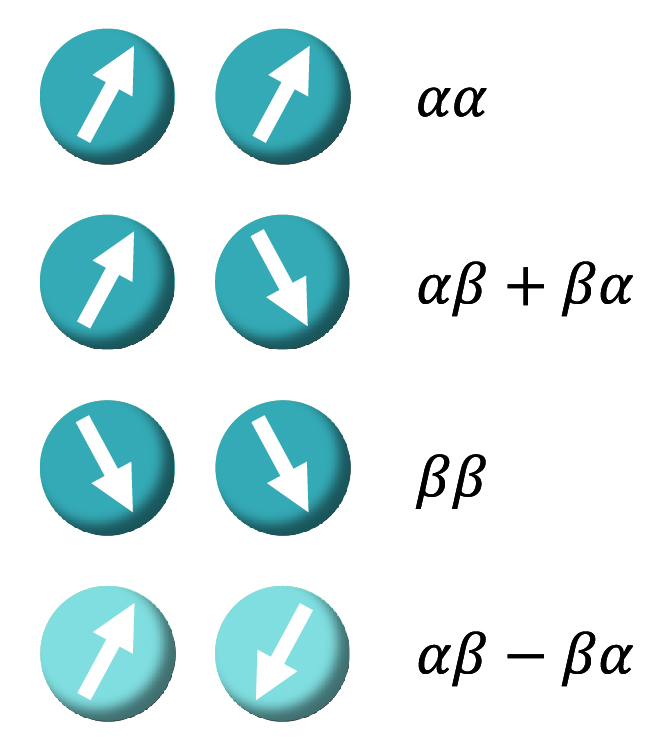
Figure 2: An illustration of the spin state combinations for molecular hydrogen.
At room temperature (T ~ 298 K), all four states are equally populated, resulting in a mixture of 25% p-H2 and 75% o-H2. Conversely, at low temperature, the para-state is much more populated as shown in Figure 3. This is because the para state is lower in energy than the ortho states due to coupling of these nuclear spin states to the molecular rotational states. The para-enrichment is obtained by flowing H2 over a catalyst (Fe2O3) at low temperature. The catalyst allows ortho→para conversion as it provides a magnetic surface for the H2 molecules to land on and convert. As can be observed in Figure 3, the enrichment is about 50% at 77 K (liquid N2 temperature), and increases to almost 100% at temperatures below 25 K.
The enrichment can be maintained at high temperature, provided that the conversion catalyst is removed, and parahydrogen can be stored in a suitable bottle/canister for months. This is of course one great advantage of the technique!
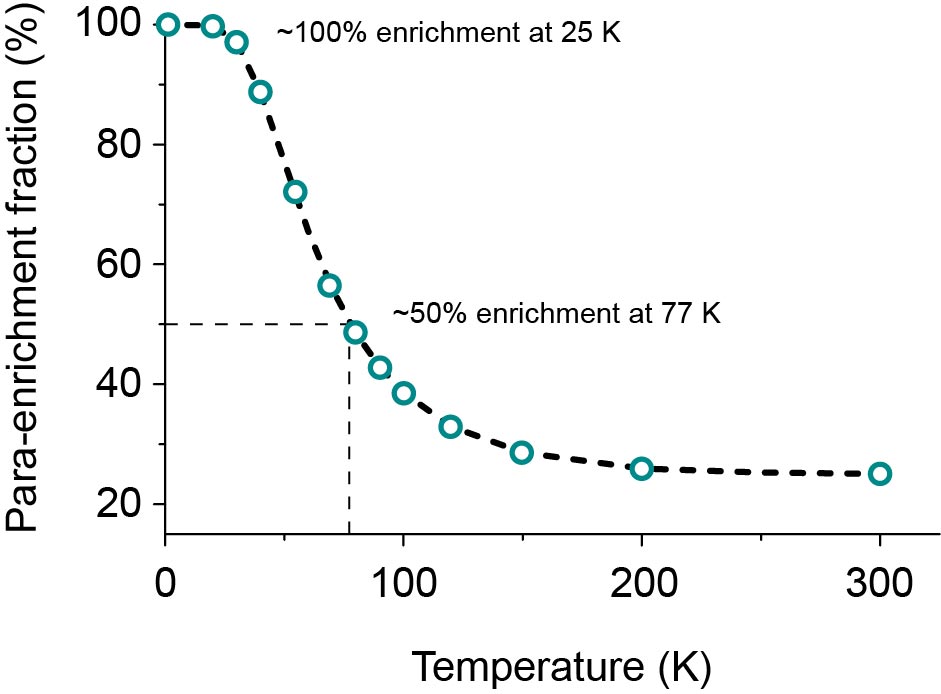
Figure 3: Parahydrogen fraction at different temperatures under thermal equilibrium conditions.
Getting enhanced signals out of parahydrogen
Parahydrogen is non-magnetic (in other words it is NMR silent), but it can generate hyperpolarization, i.e., hugely enhanced NMR signals. How is this possible?
First of all, it is necessary that the symmetry of the hydrogen molecule is broken. This occurs when parahydrogen is added to a substrate through a hydrogenation reaction. When the two protons end up in chemically (or magnetically) different positions in the product molecule and the symmetry of the H2 molecule is lost, only the states that overlap with the para-spin state are populated, thanks to the original para-enrichment. As a consequence, the transitions from these states to the other states are hyperpolarized due to the large population difference [1].
For simplicity, let us assume that the hydrogenation substrate does not contain protons and the two atoms originating from parahydrogen form an isolated two-spin system on the product molecule (Fig. 4). If the two protons have different chemical shifts we can refer to this as an AX spin system. Of the newly-formed four spin states, only αA βX and αA βX overlap with the parahydrogen singlet state, and so these two states are overpopulated. As a consequence, the transitions from these two states to the other states αA αX and βA βX are hyperpolarized and we expect to observe two antiphase hyperpolarized signals in the 1H-NMR spectrum [2,3].
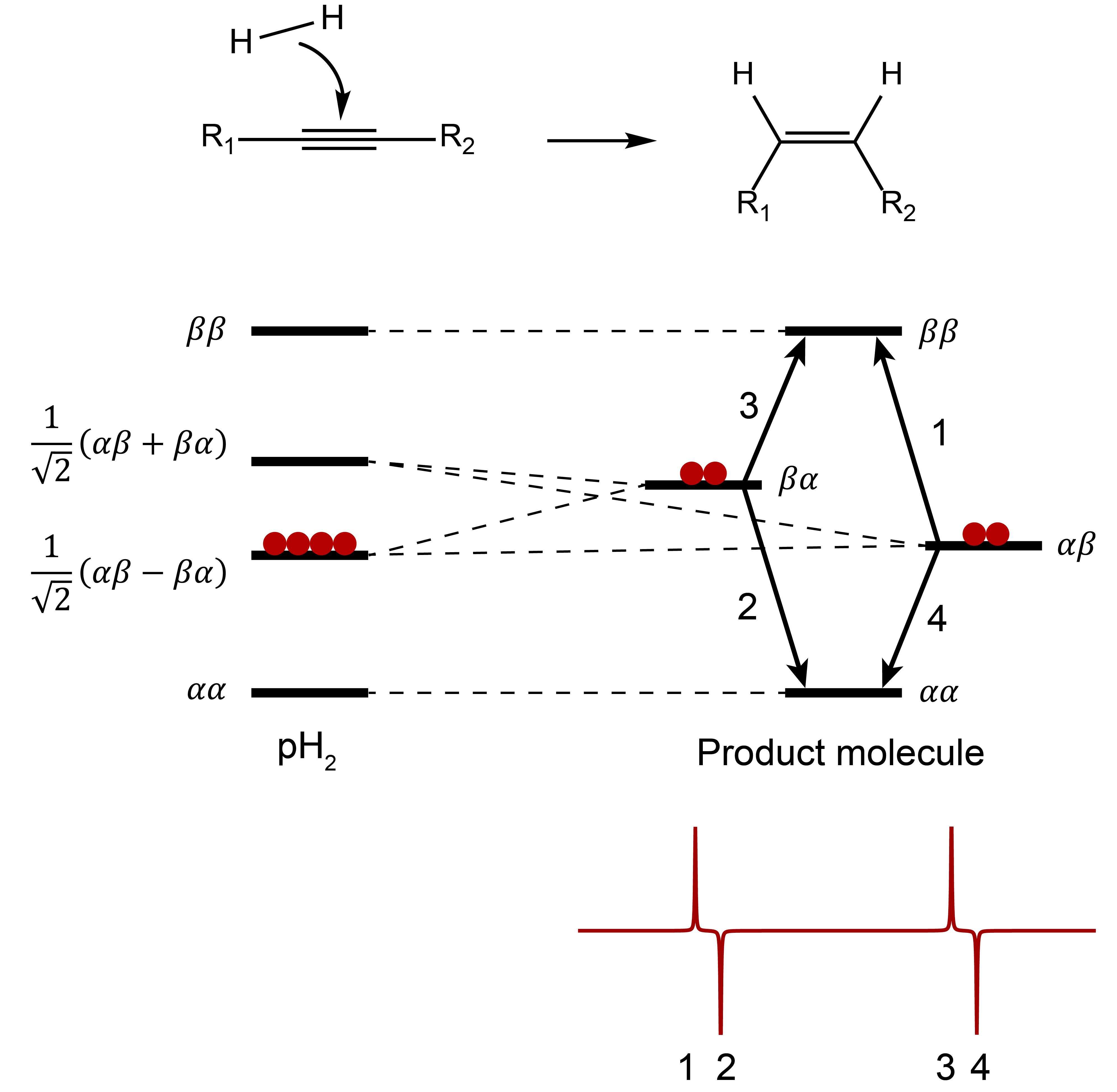
Figure 4: Schematic showing a PASADENA experiment. After a chemical reaction the parahydrogen protons end up in chemically different positions and an AX spin system is formed. This leads to an overpopulation of the αβ and βα states.
We have mentioned that the lifetime of parahydrogen can be months in the gas phase, and even once the parahydrogen is dissolved into a solution the singlet lifetime can easily be many minutes. However, once reacted, the proton hyperpolarization typically relaxes in seconds. Therefore, the rate of reaction should be high, which is possible using highly active transition metal catalysts (typically rhodium-based). Another important consideration is that the two protons have to be added to the same substrate molecule, i.e. in a pairwise manner, so that the singlet state does not get scrambled. The high pairwise addition selectivity of the PHIP catalysts is due to the specific hydrogenation pathway that they allow.
Beyond proton polarization
If the system is hyperpolarized it is by definition not in a stable state, and equilibrium is restored at a rate dictated by relaxation processes. The hyperpolarized protons would yield the highest NMR signals, but a big disadvantage are the short T1 times (in the range of few seconds). It is often worthwhile to make a trade-off by transferring the polarization to a heteronucleus in the molecule such as a 13C or 15N: the sensitivity is lower, but the hyperpolarization lifetime can be minutes. This allows the hyperpolarized molecules to be used for a much wider range of applications, including as probes in biological systems.
To efficiently transfer the proton hyperpolarization onto a heteronucleus, different methods are at our disposal. If the sample is at high field we can use rf pulse sequences, or alternatively we can utilize level anti crossings at ultralow magnetic fields for the job.
PHIP is inexpensive, it is not a technically demanding method, and the sample turnover rate can be high since the chemical reactions are fast and scalable. In spite of this there is a big drawback: the specificity of the molecules that can be hyperpolarized generally need to be produced by hydrogenation reactions. In an upcoming blog post we will explore nonhydrogenative PHIP which has helped to expand the pool of molecules that can be polarized with parahydrogen. As for hydrogenative PHIP, a particularly revolutionary new method has emerged: side-arm hydrogenation.
Extending the scope of PHIP
The side-arm hydrogenation (SAH) strategy involves bringing parahydrogen into a molecule by addition to an unsaturated side-arm moiety, which is followed by polarization transfer onto the desired target part of the molecule, followed by chemical cleavage of the side-arm [4]. A SAH procedure to generate hyperpolarized [1-13C]pyruvate is shown in Figure 5. Parahydrogen is added in a pairwise manner to the ester (propargylic or vinylic) in an organic, hydrophobic solvent (CDCl3). After polarization transfer to the 13C carboxylate site, the molecule is hydrolyzed using an aqueous base. The pyruvate migrates into the aqueous phase, and the catalyst and side-arm molecules remain in the organic phase. Hence, the phase extraction leads to a pure aqueous solution of the hyperpolarized product, which can be directly used for in vitro [5] or in vivo [6] studies.
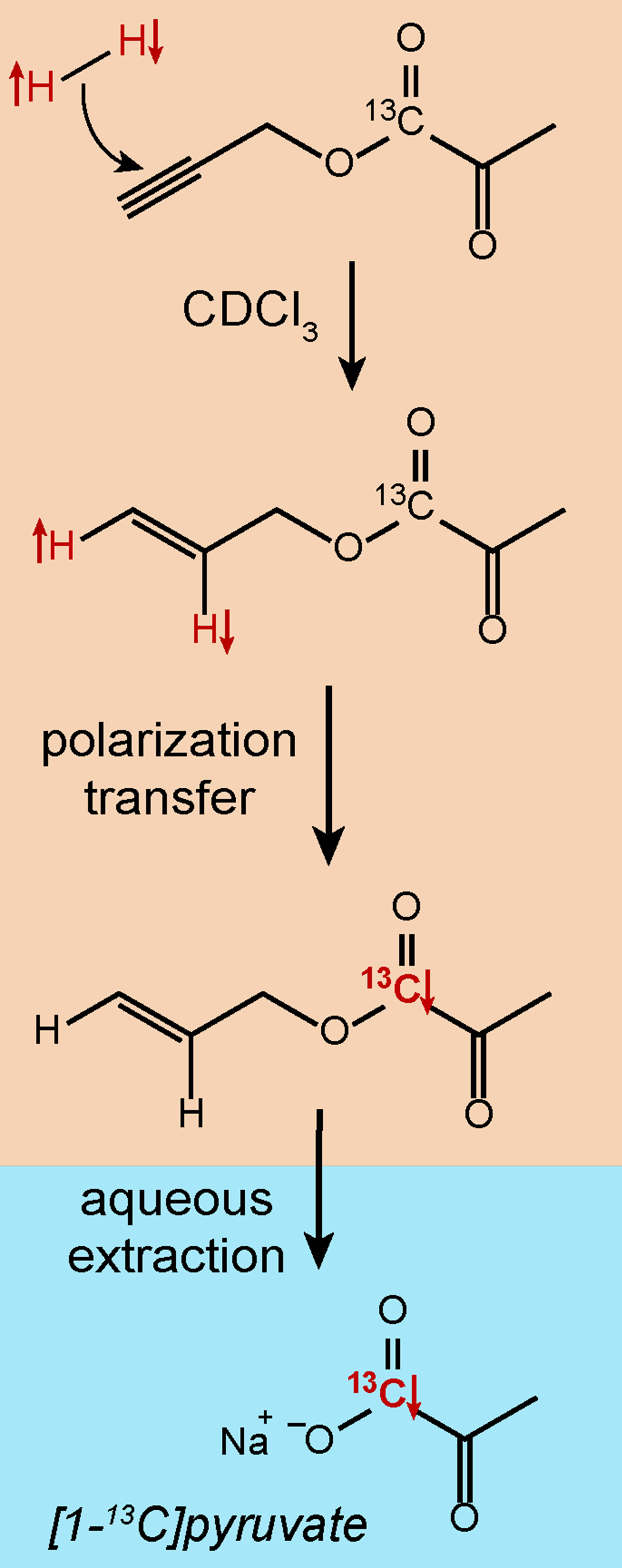
Figure 5: An example of a side-arm hydrogenation procedure to produce hyperpolarized pyruvate.
PHIP-SAH is a versatile method that greatly expands the number of molecules that can be polarized. This is highly promising for ZULF NMR [7,8], since PHIP is a natural friend: both methods are easily portable, low cost, and neither requires superconducting magnets. Let us see what the future brings!

Oksana Bondar
I am an Early stage researcher (PhD student) at the Molecular Imaging Centre Dept. of Molecular Biotechnology and Health Science University of Torino in Silvio Amie’s group, Scientific supervisor -Dr. Francesca Reineri. My scientific experience is organic and inorganic synthesis. My current project is devoted to the improving of the hyperpolarization method named PHIP-SAH (ParaHydrogen Induced Polarization- Side Arm Hydrogenation) and on the ZULF NMR detection of hyperpolarized molecule of metabolites obtained by PHIP-SAH method.
Literature references:
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler, A. L. Balch, J. Am. Chem. Soc. 109, 8089-8091 (1987)
- C. R. Bowers, D. P. Weitekamp, Phys. Rev. Lett. 57, 2645-2648 (1986)
- C. R. Bowers, D. P. Weitekamp, J. Am. Chem. Soc. 109, 5541-5542 (1987)
- F. Reineri, T. Boi, S. Aime, Nat. Commun. 6, 5858 (2015)
- E. Cavallari, C. Carrera, G. Di Matteo, O. Bondar, S. Aime, F. Reineri, Front. Oncol. 10, 1–9 (2020)
- E. Cavallari, C. Carrera, M. Sorge, G. Bonne, A. Muchir, S. Aime, F. Reineri, Sci. Rep. 8, 8366 (2018)
- J. W. Blanchard, D. Budker. Zero-to ultralow-field NMR. eMagRes. 5(3), 1395–410 (2016)
- T. Theis, P. Ganssle, G. Kervern, S. Knappe, J. Kitching, M. P. Ledbetter, D. Budker and A. Pines, Nat. Phys. 7, 571-575 (2011)

















